Abstract
The synthesis of ortho-substituted benzoic acid 3 from benzalphthalide 1 in the presence of hydriodic acid-red phosphorus (HIaq/Pred) occurs via the formation of benzylphthalide 2 as reaction intermediate. lts preparation is optimized by use of the supported reaction in "dry media" process (Silica/HIaq) under microwave irradiation for 8 min. The role of the red phosphorus in the reduction reaction is elucidated: it intervenes in a catalytic cycle by an oxido-reductive disproportionation with the liberated iodine, affording either hypophosphorous acid in aqueous media, or P2I4 in anhydrous media with concomitant regeneration of HI. Thus, the presence of Pred in the couple 'HIaq/Pred' allows recycling of the hydriodic acid and enhances its reducing efficiency.
1. Introduction
Since the couple hydriodic acid/red phosphorus (HIaq/Pred) was introduced as a reducing reagent into organic chemistry a long time ago, the synthetic aspects of its use (hydrogenation, deoxygenation of alcohols, ketones, ketoacids and quinones, cleavage of phenol ethers, and reductive cleavage of lactones) have been extensively studied1. However, the mechanistic aspects have not been thoroughly investigated, and consequently the role of the red phosphorus remains ambiguous despite its proved efficiency and its usefulness.
Previous work in our laboratory has focused on the study of the reactivity of the amorphous red phosphorus2. We have demonstrated that its basic hydrolysis allows the generation of the phosphine PH3, the hydrogen and the hypophosphite ion. Furthermore, since 1962, it was established that in acid-catalysed disproportionation, white phosphorus is converted into phosphine and phosphoric acid at high temperature3. White phosphorus, phosphine, and hypophosphorous acid are well known for their reducing properties, and the question of the role of red phosphorus in acidic media and particularly in the couple 'HIaq/Pred' arises. Pursuing our investigation into the reactivity and the reducing character of red phosphorus, we wish to report here the synthesis of the 2-(2-phenyl-ethyl)-benzoic acid 3, precursor of neurotropic derivatives, by reductive cleavage of benzalphthalide 1 in the presence of hydriodic acid-red phosphorus (HIaq/Pred), and the role of red phosphorus upon the reducing properties of this reagent.
2. Results and discussion
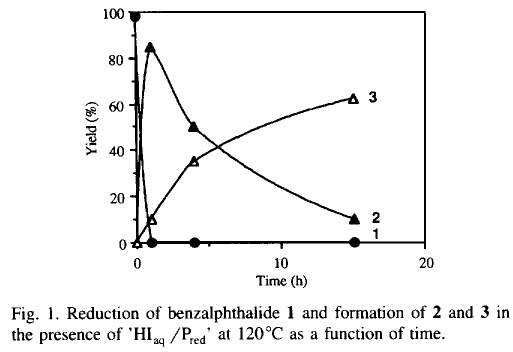
Table 1. [Enlarge]
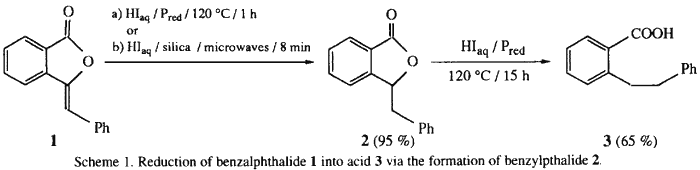
The synthesis of ortho-substituted benzoic acid 3 in the presence of hydriodic acid-red phosphorus (HIaq/Pred) requires long reaction times (15 h) under heating at 120°C (Scheme 1)4. In an attempt to improve the reaction conditions, we have realized various experiments at 120°C for the reaction times ranging between 1 h to 15 h (Fig. 1).
We observe the disappearance of benzalphthalide 1 after 1 h and the quasi-quantitative formation of a new compound which disappears as a function of time in favour of 3 (Table 1).
We have isolated this reaction intermediate 2 and its spectroscopic data are in agreement with the structure of the 3-benzylphthalide. The methods described for the preparation of 2 involve either an electrolytic reduction5 or a catalytic hydrogenation6 over Pd/C or in the presence of Raney nickel with long reaction times and low to moderate yields. We have carried out the reduction of 1 on silica in the absence of solvent ('dry media' process7) and in the absence of red phosphorus.
A solution of HIaq and benzalphthalide 1 is impregnated on silica and heated at 120°C for 1 h. The extraction of solid support by toluene allows one to obtain benzylphthalide 2 in 95% yield. When a microwave oven8 is used instead of the oil bath, the reaction time is reduced to 8 min (Table 1, exp. no. 9). Thus, this easy and selective hydrogenation of the C=C double bond by impregnated HIaq on silica constitutes a highly useful synthesis procedure, since it allows one to avoid the use of low-valent transition metal ions (Cr, Ti)9, or triethylsilane in trifluoroacetic acid10. Furthermore, it must be noted that, in our conditions, we have never observed any hydroiodination reaction11.
Since it is well known that ultrasound is more effective in the case of heterogeneous solid-liquid reactions12, we then attempted the reduction of benzalphthalide 1 in the presence of 'HIaq/Pred' under sonication at 20 kHz. When the sonication was carried out at 30°C or 80°C, no reaction occurs. However, the reaction did take place at 120°C under sonication, but without any improvement with respect to standard procedure. This is probably due to the high reaction temperature which decreases the ultrasound effects.
Consequently, the synthesis of 3 was carried out in two steps: (i) the fast hydrogenation of benzylidene moiety without ring-opening of the γ-lactone, followed by (ii) the slow hydrolysis to the benzylic cation followed by reduction. We have demonstrated that the achievement of the reaction, whether from benzalphthalide 1 or benzylphthalide 2, requires 15 h under classical heating at 120°C to afford 3 in 65% yield.
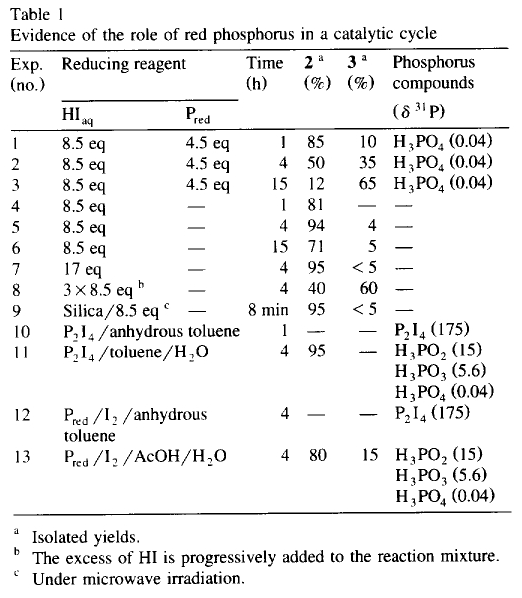
In an attempt to clarify the role of the red phosphorus in acidic media, a series of reactions were undertaken in the presence and in the absence of red phosphorus for 1, 4, and 15 h at 120°C (Table 1, exp. nos. 1-6). Whereas the reaction of 1 with 'HIaq/Pred' affords 3 in increasing yields as a function of time, the same reaction without red phosphorus is stopped at the stage of formation of 2. The 31P-NMR analysis of the reaction mixture in the presence of 'HIaq/Pred' does not exhibit the formation of organophosphorus compounds but the presence of phosphorus acids (H3PO2, H3PO3 and H3PO4). Consequently, we have assumed the presence of a catalytic cycle in which the red intervenes in an oxido-reductive disproportionation with the liberated iodine.
Actually, hydriodic acid dissociates by a reversible reaction at high temperature (120°C) to iodine and hydrogen which is the reducing reagent. In the literature, the direct generation of PH3 from white phosphorus can be realized either in acidic media at 280°C3 or by an electrolytic method13. In our conditions, the red phosphorus cannot give the phosphine PH3 directly [ E°(I2/I-) = +0.53 V and E°(Pred/PH3) = -0.111 V ]14. However, from the moment that the iodine is liberated, it reacts with the red phosphorus and affords the hypophosphorous acid and HI [ E°(H3PO2/Pred) = -0.365V ]14, allowing the equilibrium to shift in favour of the generation of hydrogen by removal of iodine and also by generation of hydriodic acid (Scheme 2). The regenerated HI functioning by a dissociative mechanism again affords hydrogen and iodine which can be consumed by red and also by hypophosphorous acid to give phosphorous acid and HI [ E°(H3PO3/H3PO2) = -0.499V ]14. Thus, the catalytic cycle can continue until the formation of phosphoric acid [ E°(H3PO4/H3PO3) = -0.276V ]14. Furthermore, this latter can be reduced by Pred to again give the hydriodic acid [ E°(H3PO4/Pred) = -0.383 V ]14 via the formation of H3PO3, etc.
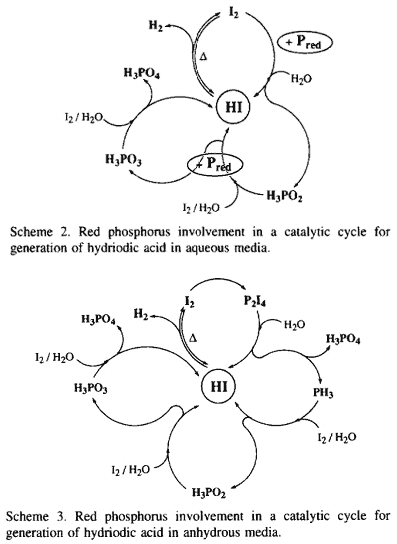
In anhydrous media, the oxidation of red phosphorus by iodine allows the equilibrium to shift in favour of the generation of hydrogen by removal of iodine as P2I4. This latter decomposes in the presence of water to phosphoric acid and phosphonium iodide which affords, under heating, the hydriodic acid and the phosphine PH315. Oxidation of PH3 by the liberated iodine in the presence of water gives H3PO2 , and the catalytic cycle can thus be continued (Scheme 3). Actually, the reduction of 2-pyrrolecarboxylic acid to the corresponding 3,4-dehydroproline in the presence of phosphonium iodide in fuming HI or aqueous solution of hypophosphorous acid saturated with gaseous HI has been realized16.
When an excess of hydriodic acid is added to the reaction mixture, we observe no improvement upon the formation of 3 (less than 5%) (Table 1, exp. no. 7).
In contrast, when the excess of HI is progressively added, 3 is obtained in 60% yield for 4h (Table 1, exp. no. 8). The role of P2I4 as reaction intermediate is demonstrated by the experiment nos. 10-13 (Table 1). Whereas the reaction of 1 and P2I4 in anhydrous toluene at 120°C for 1 h affords neither 2 nor 3, the same reaction in the presence of water (five equivalents) gives quantitatively 2 and the various phosphorus acids. The P2I4 can be generated in situ from red phosphorus and iodine17 in anhydrous toluene, and when the reaction occurs in the presence of Pred and iodine in acetic acid and water (five equivalents) at 120°C for 4 h, 2 and 3 are obtained in 80% and 15% yield respectively.
In conclusion, HIaq/Pred can be replaced by red phosphorus, iodine and water. The red phosphorus intervenes in the reducing properties of HIaq/Pred by its reducing character to consume the liberated iodine and affording either hypophosphorous acid in aqueous media, or P2I4 in anhydrous media with concomitant regeneration of HI. Thus, the driving force of the reaction is presumably the oxidation of the red phosphorus by iodine and the presence of Pred in the couple 'HIaq/Pred' allows recycling of the hydriodic acid and enhances its reducing efficiency.
Experimental
Benzalphthalide was prepared by condensation of benzylic acid and phthalic anhydride at 230°C according to the procedure already described18. Red phosphorus (Prolabo), silica (60-Merck), hydriodic acid (aqueous 57% solution) and iodine (Aldrich) were used as received.
General procedure
3.1 Conventional heating
Benzalphthalide 1 (0.2 g; 0.9 mmol) and amorphous red phosphorus (0.12 g; 3.87 mmol) were placed in the reactor fitted with a reflux condenser. A solution of aqueous 57% hydriodic acid (1 ml; 7.65 mmol) was added dropwise to the mixture then heated in an oil bath equipped with a thermostat at 120°C for 1, 4 or 15 h.
The mixture was then cooled to room temperature, a solution of toluene/ethanol (80:20) was added and the unreacted red phosphorus was filtered off. The 31P-NMR analysis of the reaction mixture exhibited the presence of hypophosphorous acid (H3PO2), phosphorous acid (H3PO3), and phosphoric acid (H3PO4).
(a) The solution was basified by an aqueous 10% solution of potassium hydroxide and extracted by toluene. The organic phase was washed with an aqueous 10% sodium hydrosulphite (NaHSO3) then with distilled water. The organic phase was dried over magnesium sulphate then evaporated to dryness. The recrystallization in hexane afforded the 3-benzylphthalide 2 as a beige solid: mp 60-61°C.
(b) The aqueous phase was acidified by an aqueous 35% hydrochloric acid and again extracted with toluene. The organic phase was washed with an aqueous 10% sodium hydrosulphite (NaHSO3) then with distilled water. The organic phase was dried over magnesium sulphate then evaporated to dryness. The recrystallization in ethanol afforded the 2-(2-phenyl-ethyl)-benzoic acid 3 as a white solid: mp 130°C.
3.2. Microwave irradiation
A mixture of aqueous 57% hydriodic acid (1 ml; 7.65 mmol) and benzalphthalide 1 (0.2g; 0.9 mmol) in dichloromethane was added to silica (1 g). The dichloromethane was then evaporated until a wet powder was obtained. This powder was then placed in a Teflon reaction vessel and irradiated in a microwave oven (Brandt ME 210 (850 W), power setting = 6) at 2450 MHz for 8 min. After cooling, the solid was extracted with a solution of toluene/ethanol (80:20), and after filtration, the mixture was treated and purified as above.
3.3. Ultrasound irradiation
Apparatus: ultrasound generator, Bioblock Vibracell 600W (20 kHz) connected to a cup horn (diam. 60 mm, power setting = 5) with a flat-bottomed flask dipped in contact with the horn. The same amounts of reagents as conventional heating were used. The reaction temperature was adjusted at 30°C, 80°C, and 120°C by means of a thermostat. The work-up was the same as that in the conventional method.
3.4. Reduction in the presence of P2I4
Distilled water (0.04 ml; 2.2 mmol) was added to a mixture of benzalphthalide 1 (0.1 g; 0.45 mmol) and P2I4 (0.25 g; 0.44 mmol) in toluene (2 ml). The mixture was heated at 120°C for 4 h, then treated as before.
3.5. Reduction in the presence of Pred/I2/H2O
To a mixture of benzalphthalide 1 (0.2 g; 0.9 mmol), red phosphorus (0.12g; 3.87 mmol), iodine (0.57g; 2.24 mmol) in glacial acetic acid (3 ml) was added 0.07 ml of distilled water (3.88 mmol). The mixture was then heated at 120°C for 4h. The work-up was the same as that above.